4. Data Extract
The module Data Extract consists of three sub-modules: EMP_assay_extract, EMP_coldta_extract, and EMP_rowdata_extract. These modules assist users in extracting specific project data from the MAE object for subsequent downstream analysis.
To help users better understand the relationships between assay, coldata, and rowdata, this section illustrates examples of species annotation (taxonomy) from 16S rRNA gene sequencing, along with corresponding annotations for microbial functional genes such as KO annotations (geno_ko) and EC enzyme annotations (geno_ec).
4.1 Extract assay (experimental data)
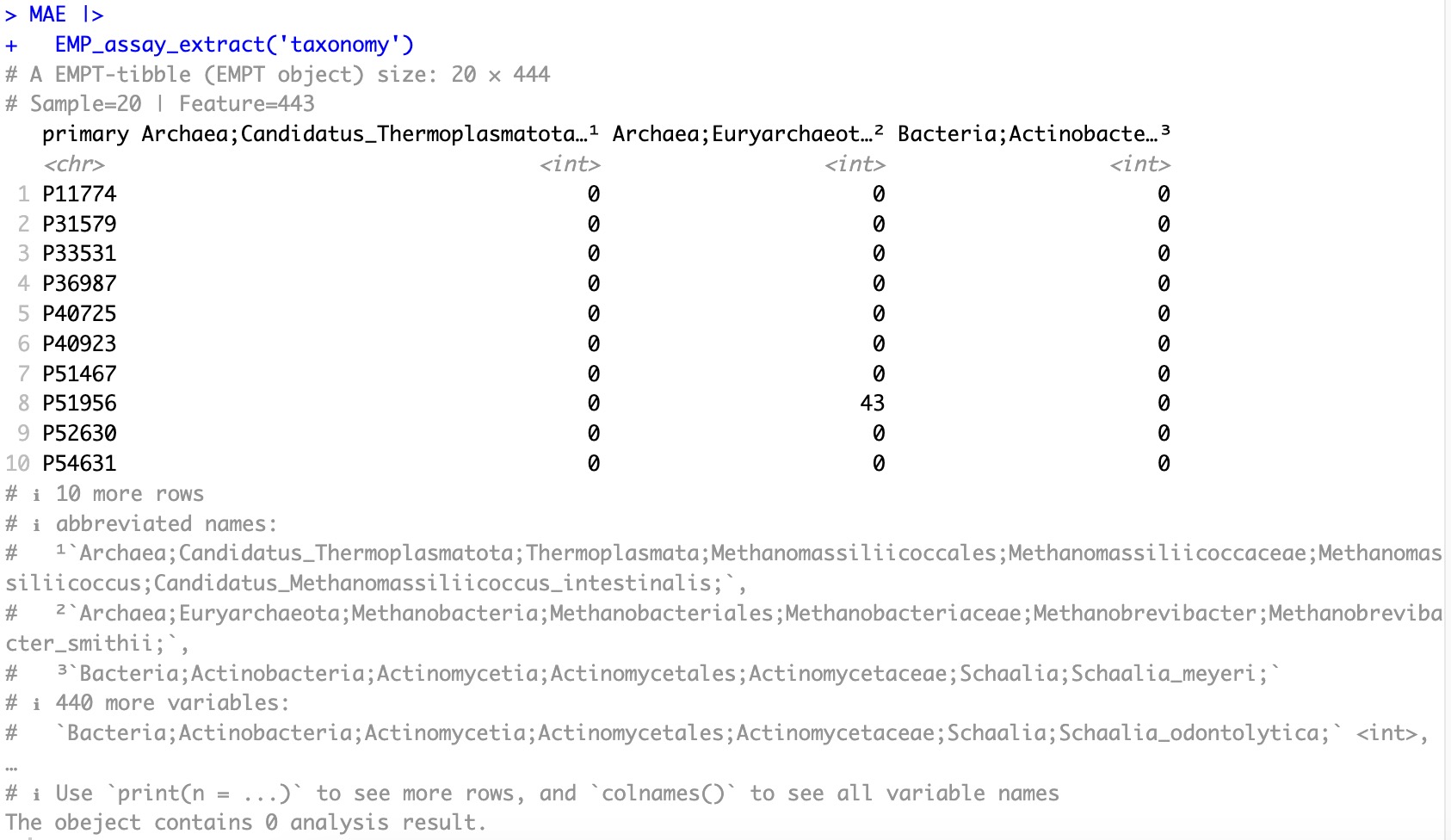
🏷️Example 1: Extract the assay of taxonomy and obtain the species annotated abundance matrix.
MAE |>
EMP_assay_extract(experiment='taxonomy')
🏷️Example 2: Extract the taxonomy assay, identify the features of Lactobacillus in the Genus column of the rowdata by specifying the parameters pattern_ref and pattern, and then extract corresponding data.
① The module
EMP_assay_extract provides internal retrieval parameters to facilitate quick discovery of abundance matrix for features of interest.② Both parameter
pattern and pattern_ref here are used for string matching and searching based on rowdata.③ Specify the parameter
action='get' to directly extract the data frame of the data matrix.
MAE |>
EMP_assay_extract(experiment='taxonomy',
pattern = 'Lactobacillus',pattern_ref = 'Genus')

🏷️Example 3: Extract the assay of geno_ko, find the puua or puuc features in the Name column of roldata by specifying the parameters pattern_ref and pattern,and further extract the KO annotation abundance matrix corresponding to these features in the assay.
Both parameter
pattern and pattern_ref here are used for string matching and searching based on rowdata.
MAE |>
EMP_assay_extract(experiment='geno_ko',
pattern = c('puua','puuc'),pattern_ref = 'Name')

4.2 Extract rowdata (feature-related data)
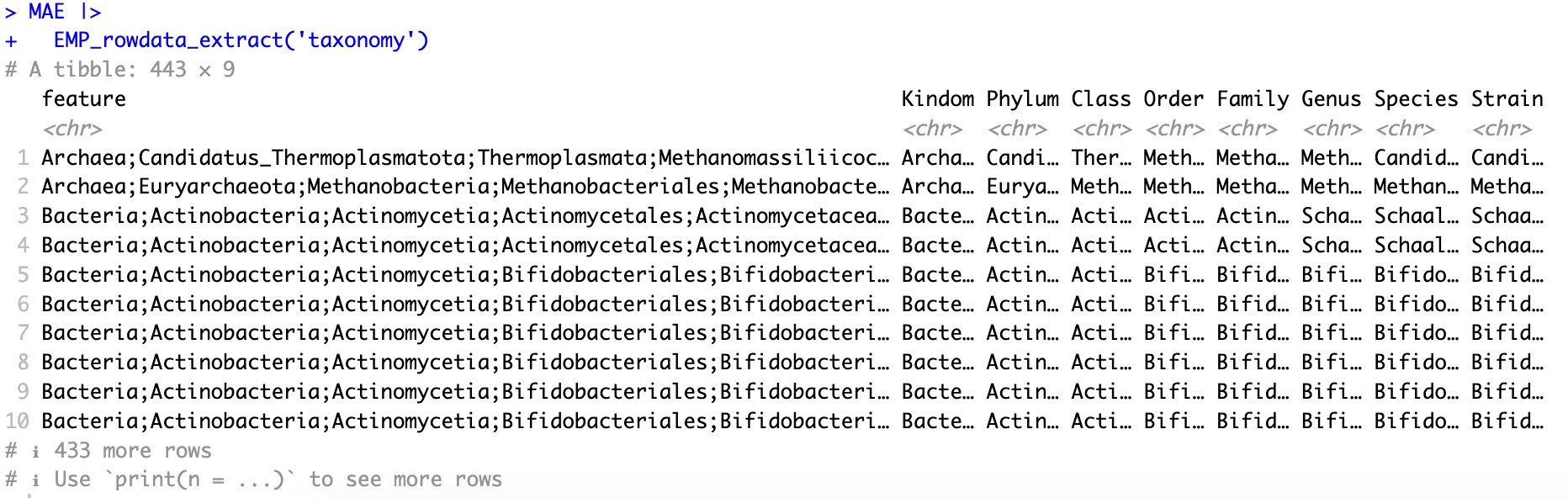
① When the module
EMP_rowdata_extract specifies a project name, the output is feature-related data of the project (For example, specifying EMP_rowdata_extract(experiment='taxonomy', outputs taxonomic annotations at the kingdom, phylum, class, order, family, genus, and species levels for the features. Specifying EMP_rowdata_extract(experiment='geno_ko') outputs relevant information from the KEGG database for corresponding KO genes. If the parameter experiment is not specified, it outputs all the feature-related data within the MAE object.② This module does not support the parameter
action.
🏷️Example 1: Extract the rowdata of taxonomy.
MAE |>
EMP_rowdata_extract(experiment='taxonomy')
🏷️Example 2: Extract the rowdata of geno_ko.
MAE |>
EMP_rowdata_extract(experiment='geno_ko')

🏷️Example 3: Extract the assay of geno_ko, find the puua features in the Name column of rowdata by specifying the parameters pattern_ref and pattern, and extract the KO annotation abundance matrix corresponding to these features in the assay. Finally, view the information related to the puua features.
MAE |>
EMP_assay_extract(experiment='geno_ko',
pattern = 'puua',pattern_ref = 'Name')|>
EMP_rowdata_extract()

4.3 Extract coldata (sample-related data)
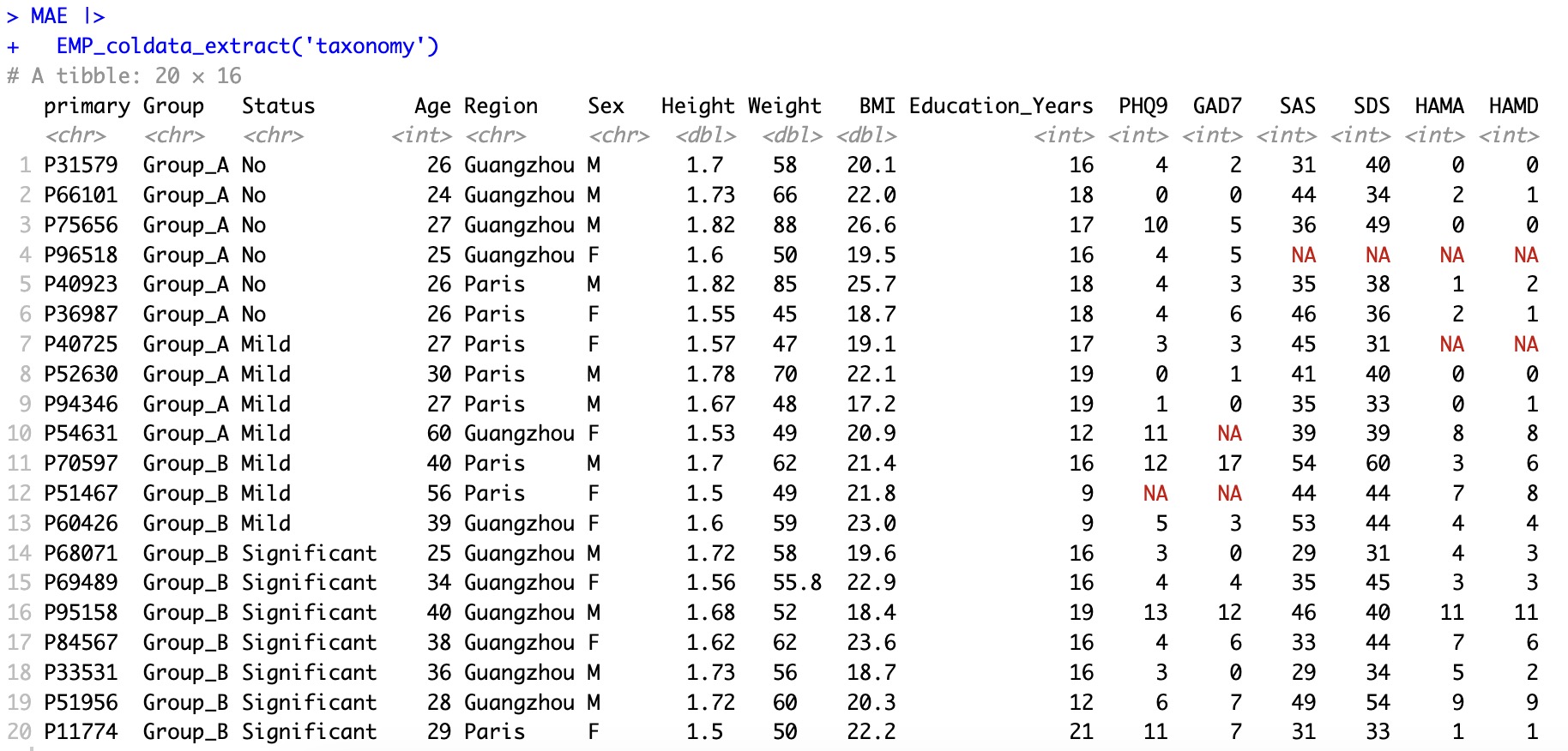
When module
EMP_coldata_extract specifies an project name internally, it outputs sample-related data for the samples of the project. If the parameter experiment is not specified, it outputs sample-related data for all samples in the entire MAE object.
🏷️Example 1: Extract the coldata of taxonomy.
MAE |>
EMP_coldata_extract(experiment='taxonomy')
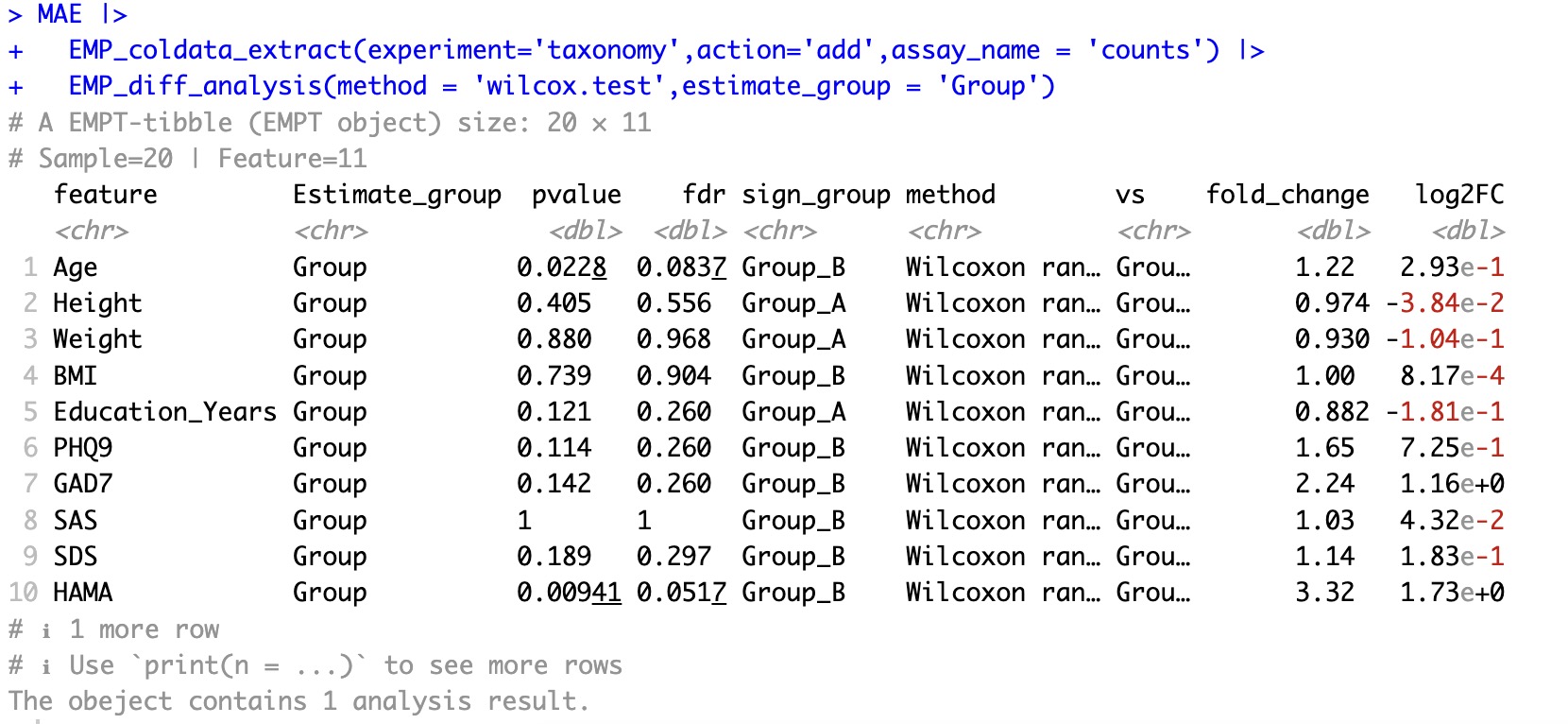
🏷️Example 2: Convert the sample related data (coldata) into experimental data (assay) and perform difference analysis.
The EasyMultiProfilerpackage uses assay by default for process profiling. If the user needs to analyse coldata, the parameteraction='add' should be specified to convert coldata to assay, and then perform various module analysis.
① In the module
EMP_coldata_extract, the parameter coldata_to_assay can specify the conversion of specific coldata to assay.② In the module
EMP_coldata_extract, when the parameter coldata_to_assay is omitted, all continuous variables in coldata are converted to assay by default.
MAE |> EMP_coldata_extract(experiment='taxonomy',action='add',assay_name = 'counts') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group')